君实生物陷舆论漩涡 “首款国产PD-1”遭质疑
来源: 华夏时报
作者:孙源 于
摘要: 因11月12日的一篇自媒体文章,而遭上交所火速问询的君实生物(1877.HK,688180.SH),科创板以及港股股价双双跳水,自12日开盘至13日15:00,科创板股价跌幅达9.98%,
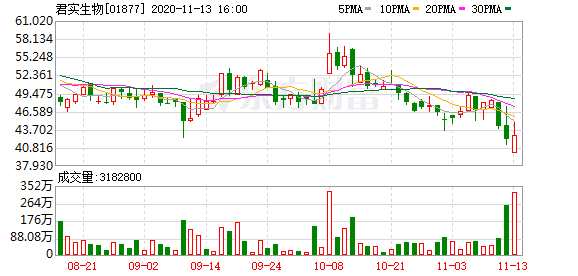
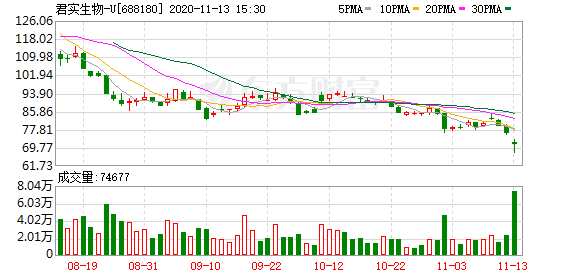
因11月12日的一篇自媒体文章,而遭上交所火速问询的君实生物(1877.HK,688180.SH),科创板以及港股股价双双跳水,自12日开盘至13日15:00,科创板股价跌幅达9.98%,港股股价跌幅达6.88%。
对于君实生物核心产品PD-1特瑞普利单抗(拓益)、研发团队以及与礼来合作的中和抗体(JS016)等方面遭遇的质疑,君实生物对该问询函的回复同样迅速,君实生物相关负责人11月13日下午对《华夏时报》记者透露,公司在13日早上已向上交所递交了对于问询函的回复。13日深夜,君实生物发布了对上交所的回复公告。
对于该事件,君实生物还未等至上交所审核发布其回复函,便发布H股澄清公告及官方公众号声明。对已于向上交所递交回复函的内容是否比目前的两则回应信息更为详细,对方回答:“详细得多,所以也会作为我们的标准回复材料。”
97.7%不良反应发生率高不高?
君实生物的特瑞普利单抗在2015年获得原国家食药监局临床批准,并在2018年获得国家药监局有条件批准上市用于治疗黑色素瘤,为第一个获批上市的国产抗PD-1单抗。
上交所指出,有微信公众号发表文章称,公司产品特瑞普利单克隆抗体注射液(拓益)“在技术评审的文件中,既没有完成肝损害患者试验、也没有完成肾损害患者试验,其所有不良反应发生率为97.7%。有15.6%的患者因为不良反应而永久停药。”
上交所要求君实生物结合临床试验数据,说明“所有不良发生率”的具体含义,核实前述报道是否属实,并补充披露特瑞普利单克隆抗体注射液临床试验的进展情况;同时结合自身产品及市场中同类产品的相关实验数据;并说明特瑞普利单克隆抗体注射液安全性和有效性。
在港股澄清公告中,君实生物罗列了旗下PD-1与其他国产PD-1产品的不良反应发生率的数据,对比来看,3家国产PD-1的安全性数据基本无差别。其中,君实生物的特瑞普利单抗所有级别的不良反应发生率为97.7%,三级及以上不良反应发生率为28.9%。信达生物的信迪利单抗所有级别的不良反应发生率为99%,三级及以上不良反应发生率为33.3%。恒瑞医药的卡瑞利珠单抗所有级别的不良反应发生率为100%,三级及以上不良反应发生率为26.7%。
实际上,临床试验方案中,一级不良反应如皮肤瘙痒、乏力、食欲下降均属于不良反应。对于肺炎、腹泻或结肠炎等涉及3级以上的不良反应,治疗调整方案为永久停药。君实生物在回复问询函时表示,作为抗肿瘤药物,一般都有较高的“所有不良反应发生率”,其中更受临床医生关注的为 3 级及以上不良反应发生率及与药物相关的严重不良反应(SAE)发生率。
由于国产PD-1有适应症上的差异,君实生物还以同适应症的国际进口PD-1资料进行了对比。在黑色素瘤的疗效上,特瑞普利单抗客观缓解率为17.3%,进口的默沙东帕博利珠单抗为16.7%,疾病控制率上,特瑞普利单抗为57.5%,帕博利珠单抗为38.2%,中位生存期特瑞普利单抗为22.2月,帕博利珠单抗为12.1月,12个月总生存率特瑞普利单抗为67.3%,帕博利珠单抗为50.6%。
君实生物同时解释,由于各公司临床试验是在不同肿瘤患者人群和不同条件下进行的,不同临床试验中观察到的不良反应的发生率不能直接比较。此外基于单臂单药的汇总安全性数据也会受到晚期恶性肿瘤自身和既往伴随用药的混杂因素影响,因此也不能直接比较。
对于拓益新适应症的开发仍在进行中。7月份,特瑞普利单抗治疗鼻咽癌、尿路上皮癌的两项新适应症上市申请获药监局优先审评,成为全球首个抗PD-1单抗治疗复发/转移性鼻咽癌的新药上市申请。目前,特瑞普利单抗正在全球开展超过30个单药治疗及联合治疗的临床试验。中信建投研报预计,君实生物在2021年、2022年将有多个新适应症上市申请,持续扩大特瑞普利单抗的适用范围。
PD-1赛道的“有条件批准”
“既没有完成肝损害患者试验、也没有完成肾损害患者试验”又作何解释?君实生物回复上交所问询表示:“从临床试验患者安全风险控制角度,中重度肝功能和/或肾功能异常患者在临床试验中不符合入组标准而没有入组临床试验,就如乳腺癌的临床研究中一般都排除了男性乳腺癌患者。”《华夏时报》记者查询,君实生物已在特瑞普利单抗注射液说明书中描述了包括肝肾损伤相关的副作用风险,提示产品在中度或重度肝、肾功能损伤患者中使用的安全性和有效性尚未建立,不推荐用于中、重度肝、肾功能损伤的患者。
君实生物表示,此类患者已作为重要的缺失患者人群纳入本品上市后风险管理计划,强调临床医生在上市后真实世界使用本品时应根据个例患者的实际情况,注意对肝肾功能不全患者的监测和管理,同时严格按《说明书》执行免疫相关器官毒性管理指南。
而拓益的快速上市,得益于“有条件批准上市”的制度。
君实生物回复上交所表示,申请有条件批准的新药应该是拟用于预防或治疗严重疾病或降低疾病进展至更严重程度的药品,包括治疗罕见病的药品。目标适应症的现有治疗手段应具有未被满足的临床需求。特瑞普利单抗在进行新药申请时,国内并无针对既往标准治疗失败后的局部进展或转移性黑色素瘤的抗PD-1单抗获批,且特瑞普利单抗与既有疗法相比对疾病的严重结果有明显改善作用,属于针对治疗严重疾病或降低疾病进展至更严重程度且具有未被满足临床需求的药品,符合有条件批准的相关条件。
在国内,除君实生物以外,信达生物的信迪利单抗、恒瑞医药卡瑞利珠单抗等国产PD-1企业均是在完成I至II期的早期临床试验后即获得有条件批准上市,上市后再向药监部门补充进行III期临床试验,并对现有试验进行继续研究,目的是为了实现国产品种的尽快上市。行业人士指出,对于强效的抗肿瘤药物来说,其发生不良反应几乎不可避免,尤其是对于面临死亡威胁的癌症晚期病人来说,其对于疗效的需求远超副作用的风险。
不只是中国,美国FDA审批“孤儿药”认定也遵循此原则。所以,凭借现有的临床试验数据,拓益也得到了FDA的认可。“今年,特瑞普利单抗在黏膜黑色素瘤、鼻咽癌、软组织肉瘤治疗领域获得美国FDA授予的3项孤儿药认定。特瑞普利单抗治疗鼻咽癌获得了美国FDA授予的突破性疗法认定。特瑞普利单抗也是中国第一个获得美国FDA突破性疗法认定的自主研发抗PD-1单抗。”君实生物13日在公众号声明中表示。
经记者向药学专业人士咨询得知,对于有条件批准上市的药物,因为其在Ⅱ期临床使用了替代终点指标,并不一定能真正验证临床的真实疗效,对于疾病转归的预测作用尚需进一步观察探索,所以在临床实践中还需继续观察远期疗效及疾病转归等。
如何确保上市后企业在规定时间内按要求完成III期确证性临床,而不是撒手不管?按照规定,对于使用早期临床试验数据申请有条件批准上市的药品,都会要求申请人应在申请上市许可时提交承诺的上市后确证性临床研究计划和方案以及完成期限、上市后风险控制计划等,否则会面临注销有条件批准药品的药品注册证书的严厉处罚。
同时,巨大的临床需求前,是国产PD-1相对进口药更加可及的价格,这也进一步扩大了患者对于该药品有条件批准上市的呼声。君实生物的PD-1拓益上市前,国内患者只有默沙东和百时美施贵宝的两款进口产品上市,费用高昂。“为了最大程度满足中国患者的可及性,公司将特瑞普利单抗的上市价格定位在同类产品海外定价的1/6水平。”君实生物在声明中表示。
如今,君实生物拓益的III期补充临床试验正在进行中,公司表示,在特瑞普利单抗有条件获批时已就要求的确证性临床试验方案与监管机构达成一致意见并开始实施,即一项考察JS001对比达卡巴嗪一线治疗不可切除的或转移性的黑色素瘤的随机、对照、多中心、III期临床研究。公司表示,需要在五年有效期内完成上述确证性临床试验。由于新冠疫情影响病人入组速度,且黑色素瘤在中国属于小适应症,发病率低,根据目前的临床进展情况,公司预计争取将于2021年底前完成该临床试验。
此外,关于自媒体对其新冠中和抗体JS016相关海外临床试验被礼来停止的质疑,根据礼来此前的对外解释与君实生物今日的澄清,该自媒体所言被暂停的试验,是评估LY-CoV555联合标准治疗(瑞德西韦)对比安慰剂联合标准治疗(瑞德西韦)的III期临床试验,而该自媒体将其中的瑞德西韦错指为JS016,JS016的相关临床试验进展并未停止。
关键词:
审核:yj115
编辑:yj127
君实生物,单抗,普利